医药
医药
现在主要的问题是人的问题,人的精神问题、身体问题。这些都和医药健康行业相关,经济可以不行,但是人不得不吃药。就算你收入减少,也会生病,也不能不吃药。经济差、收入少,情绪低落,更加容易生病。医药行业,比白酒和科技更加稳健。白酒其实对身体无益,可以不喝;科技变得太快了,太虚无了,也不实际。看来还是医药保险。
医药行业太复杂了,涉及公司很多。一般细分行业大概分为六大类:
化学制药、生物制药、中药、医药商业、医疗器械、医疗服务。
也就是有:有制药的,有买药的;有中药的,有西药的;有生产的,有服务的;有治病器械的,有医用耗材的;有治病的,有美容的;还有各种的医学分科,根据系统功能上分科的内科(神经、心血管、呼吸、消化、内分泌、风湿免疫、肝脏等)、根据解剖结构上分科的外科(普通、心胸、泌尿、骨、脑、烧伤整形;按疾病性质:肿瘤、微显、急诊、创伤等),还有独特人群的妇产科和儿科。
fast follow、license in、first-in-class/best-in-class
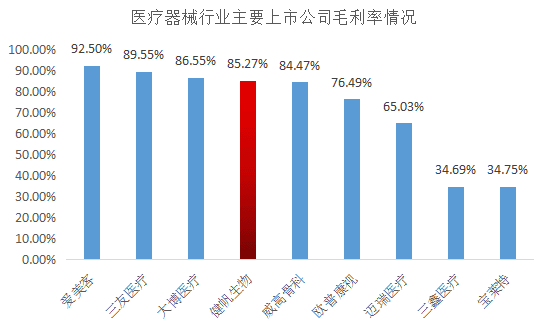
抗癌药 | CXO | 港股B | 医美 | 疫苗 | 骨科 | Diagnosis
国家医保服务平台:药品目录 | 国家药品监督管理局:药品评审中心
透视国家医保目录准入调整的背后逻辑!
阿里、腾讯等巨头入局医疗 背后商业逻辑各不同
2019年最全解析“抗癌神药PD-1”
肺癌如何选择PD-1/PD-L1药物?这里有一份最新指南
2020医保谈判启动 6家PD-1产品上场角逐
以往疫苗从研发到上市要经历10-15年,每种成功的疫苗几乎使用过当时所有的研发手段,但是最终研发成功的疫苗并不多。全球目前只有约70种疫苗,用于预防37种传染病。
疫苗研发和药物研发不一样,疫苗是给健康人群用的,研发的难度更大。更困难的是,疫苗要先在老鼠身上做试验。但目前的新冠肺炎病毒无法通过老鼠ACE2蛋白入侵,这时候就要用转基因技术把人ACE2蛋白的基因转到老鼠体内。而用于试验的老鼠,从怀孕到生下小老鼠,再到小鼠长成具有免疫功能的老鼠,大概需要近一个月的时间。除了老鼠外,疫苗还要在非人灵长类动物身上做实验。除此之外,还要经过毒理实验、动物实验、临床试验才算走完疫苗研发的整个流程。一切顺利的话,疫苗也要一年时间研制出来。
传统疫苗,比如灭活疫苗;减毒活疫苗;现代疫苗,重组蛋白疫苗(赛诺菲与葛兰素史克);核酸疫苗(MRNA,辉瑞与德国BioNTech,法国赛诺菲与美国TranslateBio);重组病毒载体疫苗(陈薇院士);
中国目前已获批进入临床试验的疫苗有三种:陈薇院士的腺病毒载体疫苗,还有两种灭活疫苗:武汉生物制品研究所和中国科学院武汉病毒研究所以及北京科兴中维生物技术有限公司。
1. 医药总龙头:600276恒瑞医药
2. 中药总龙头:000538云南白药
3. 新药实验龙头:603259药明康德
4. 糖尿病龙头: 600867 通化东宝
5. 大输液龙头: 002422科伦药业
6. 维生素用药龙头:002001新和成
7. 血制品龙头: 002007华兰生物
8. 生长激素龙头:000661长春高新
9. 中枢神经龙头:002262恩华药业
10.眼科服务龙头:300015爱尔眼科
11.牙科服务龙头:600763通策医疗
12.膏贴药龙头:002287奇正藏药
13.核药龙头:002675东诚药业
14.医药商业龙头:601607上海医药
15.家用医疗器械龙头:002223鱼跃医疗
16.骨科材料龙头:002901大博医疗
17.心脏支架龙头:300003乐普医疗
18.药用玻璃龙头:600529山东药玻
19.疫苗龙头:300601康泰生物
20.临床CRO龙头:300347泰格医药、
21.独立实验室龙头:603882金域医学
22.体检业务龙头:002044美年健康
23.化学发光诊断试剂龙头:603658安图生物
24.血液灌流器械龙头:300529健帆生物
25.药店龙头:603939益丰药房
26.抗过敏生物药龙头:300357我武生物
图片来源:虎嗅
医疗服务
临床试验:药明康德、泰格医药、凯莱英、昭衍新药
医疗器械:迈瑞医疗
IVD体外检测:金域医学、迪安诊断、科华生物、安图生物、达安基因、华大基因
医疗信息化:卫宁健康、创业惠康、思创医惠
20200204 第一财经 李家琪
阿尔茨海默病,就是老年痴呆症。我国第一款治疗阿尔茨海默症(AD)的原创新药——甘露特钠胶囊(商品名“九期一”)近日有条件获批上市,填补了该领域全球17年无新药上市的空白。“九期一”是我国原创、国际首个靶向脑—肠轴的阿尔茨海默症治疗新药,其研发逻辑背后,是对阿尔茨海默症发病机理的一种全新认识。
据了解,有12家制药公司获得生产双黄连口服液的相关许可,目前涉及双黄连口服液的上市公司共5家,包括港股的福森药业,A股的哈药股份、太龙药业、珍宝岛,以及天士力持股45%的河南天地药业股份有限公司。
2月3日,除了天士力之外,哈药股份、太龙药业、珍宝岛涨停,港股的福森药业甚至一度暴涨逾200%,最高见20港元。
资料显示,福森药业的运营主体为河南福森药业有限公司,成立于2003年10月,注册资本约7676万人民币,法定代表人为董事长曹长城。公司经营范围包括生产:片剂、硬胶囊剂、颗粒剂、小容量注射剂、大容量注射剂、口服液、原料药。
可以看到,福森药业在1月份每日成交量都很低,正常维持在几十万元交易额,甚至最低一日只有8万元,属于流动性很差的个股。但一旦遇到概念爆炒2月3日成交额超亿元。在市场普跌的情况下,资金抓住医疗板块的概念炒作一下,是正常的,但像福森药业这般就要注意了。
果不其然,暴涨200%后,福森药业的股价快速回落,截至午盘涨幅缩窄至53.14%。
同时,福森药业发公告称,截至本公告日期,本集团尚未就双黄连口服液抑制新型冠状病毒的功效开展任何研究活动,亦未就有关功效作出任何声明及保证。

IND、NDA、ANDA
FDA(Food and Drug Administration)新药审评程序包括新药临床试验申请IND(Investigational New Drug)申报和新药申请NDA(New Drug Application)申报两个过程,申请人在完成新药临床前研究后,便可向FDA提出IND申请,若FDA在收到后30天内未提出反对意见,申请人便可自行开展新药临床研究。仿制药申请通常被认为是简短的,因为这类申请不需要提供临床前动物和临床人体数据来证明其安全性和有效性。下面来看一下什么是IND申报、NDA申报和ANDA申报?
1、IND申报
IND主要目的是提供足够信息来证明药品在人体进行试验是安全的,以及证明针对研究目的的临床方案设计是合理的。IND主要包括Ⅰ、Ⅱ、Ⅲ期临床试验申请,其中Ⅰ、Ⅱ期临床试验为初期临床试验,是疗效的探索阶段;Ⅲ期临床试验为扩大临床试验,是疗效的验证性阶段,只有在初期临床试验IND获准后,申请人才可以提交扩大临床试验申请。
在IND申报阶段,FDA一般规定(最低限度)药品申办者必须:(1)做该药的药理研究;(2)在至少二种动物身上进行急毒试验;(3)按照该药预想的用途进行为期二个星期至三个月的短期研究。一旦临床前研究结束,动物试验并没有结束随之完成,许多时间更长、更专项的研究如慢性、抗癌试验将在整个新药申请过程中进行。
临床前研究用来评估: (1)药品的药理学现象和作用机理(MOA);(2)药物毒性特征和毒性靶器官;(3)药物吸收、分布、代谢和排泄(ADME)。当药品申办者认为它已具有足够的数据证明该药是安全时,就可准备向FDA提交新药临床研究申请(IND)。本质上IND只是一个建议,通过这个建议,药品申办者获得FDA的许可,开始在人身上进行试验。
2、NDA申报
当人体试验第三阶段完成,所需非临床试验已告结束,则可以出具一套资料,向FNS申请新药上市许可之核准,NDA主要目的是确保上市药品安全有效和质量可控。以植物药NDA为例,FDA认为除其在质量标准方面与化学药品确有不同之外,在安全性和有效性方面二者应符合相同上市标准。因此FND对植物药NDA在安全性和有效性方面的审批要求与相同适应证的化学药品相同,但在质量标准方面可有所不同。
植物药NDA申报材料与化学药品类似,主要包括以下内容:CMC数据、非临床药理和毒理数据、人体药代动力学和生物利用度数据、微生物数据、临床数据、安全性数据更新报告、统计学数据、病例报告表、有关专利情况、样品、包装及标签等。
一般符合以下情况均可向FDA提出NDA申请:
(1)新分子实体 (NME);
(2)新化学实体(NCE);
(3)原批准药品相同化学成分的新盐基、新酯基;
(4)原批准药品的新配方组成;
(5)原批准药品的新适应症(包括处方药转非处方药使用);
(6)新剂型、新给药途径、新规格(单位含量);
(7)两种以上原批准药品的新组合。
3、ANDA申报
ANDA的申请即为“复制”一个已被批准上市的产品。其中,这里的“复制”是指其与该上市药品具有相同的活性成分、剂型、规格、服用方式及适应症等。仿制药申请被称为简短的 (abbreviated),是因为这类申请不需要提供临床前(动物)和临床(人体)数据来证明其安全性和有效性。取而代之的是,仿制药申请者必需提供产品生物等效性的证明材料(比如与原研药相比没有区别)。一旦此类药品获得批准,申请者可以生产并上市这一安全有效且价格低廉的替代物。
BE试验是为了比较仿制药和原研药体内吸收程度和吸收速率,因此提供源于BE试验的数据是ANDA申报资料中一个关键的组成部分。与评估药学等效性一起,确立生物等效性允许在药政监管层面上做出治疗等效性的结论。
不可或缺的铲子
血制品上市公司简析-上海莱士、华兰生物
上海莱士VS华兰生物:炒股的输给了搞疫苗、单抗的
一文读懂CRO四家上市公司:药明康德、康龙化成、泰格医药、凯莱英
论复星医药与恒瑞医药的两种模式
年报分析:恒瑞医药VS复星医药,殊途同归未了局
“药王是怎样炼成的”
红杉、高瓴的医药布局逻辑
恒瑞医药进军眼科 A股还有哪些值得关注的眼科公司